
La mucoviscidose est la plus fréquente des maladies génétiques rares chez les caucasiens d’Europe et d’Amérique du Nord. Elle est causée par une mutation du gène CFTR qui code pour la protéine Cystic Fibrostic Transmembrane conductance Regulator présente dans la membrane des cellules des muqueuses des organes creux. C’est cette protéine qui donne au mucus sa fluidité ; lorsqu’elle est altérée, le mucus de l’individu malade est plus abondant et plus visqueux. À ce jour, il n’existe pas de moyen de guérir la maladie mais les traitements palliatifs sont nombreux, permettant ainsi aux malades de vivre plus longtemps.
La mucoviscidose fait partie des maladies diagnostiquées dès la naissance selon le protocole établi en 2002. Dans un premier temps, on évalue le taux de trypsine dans le sang du nouveau-né âgé de trois jours. Si celui-ci est élevé il y a suspicion de mucoviscidose, on analyse alors dans un second temps le même échantillon sanguin pour y déceler des mutations de la protéine CFTR. Dans le cas où des altérations sont trouvées, on réalise le test « de la sueur » pour évaluer la concentration en ions chlorure de la sudation du bébé qui est très importante chez les malades.
La mucoviscidose est autosomique récessive : elle s’exprime donc quand les deux allèles portés par la paire de chromosomes 7 sont mutés. Lorsque seulement l’un des allèles est muté, l’individu est considéré comme « porteur sain » et a alors une chance sur deux de le transmettre à ses enfants.
Par le passé, la recherche a permis d’identifier 2.000 altérations possibles de ce gène. Chaque altération provoque des symptômes plus ou moins graves et donc une forme de la maladie plus ou moins importante. Cependant, la mutation la plus fréquente est la mutation Delta F508, elle entraîne d’ailleurs une forme assez sévère de la maladie. Elle est présente chez 70% des malades hétérozygotes et 50% des homozygotes.
La protéine CFTR permet un échange d’ions chlorure entre les espaces inter- et extracellulaires. Lorsqu’elle présente des anomalies, le canal associé ne fonctionne plus correctement : les muqueuses s’assèchent, le mucus devient plus épais, la mucoviscidose se manifeste.

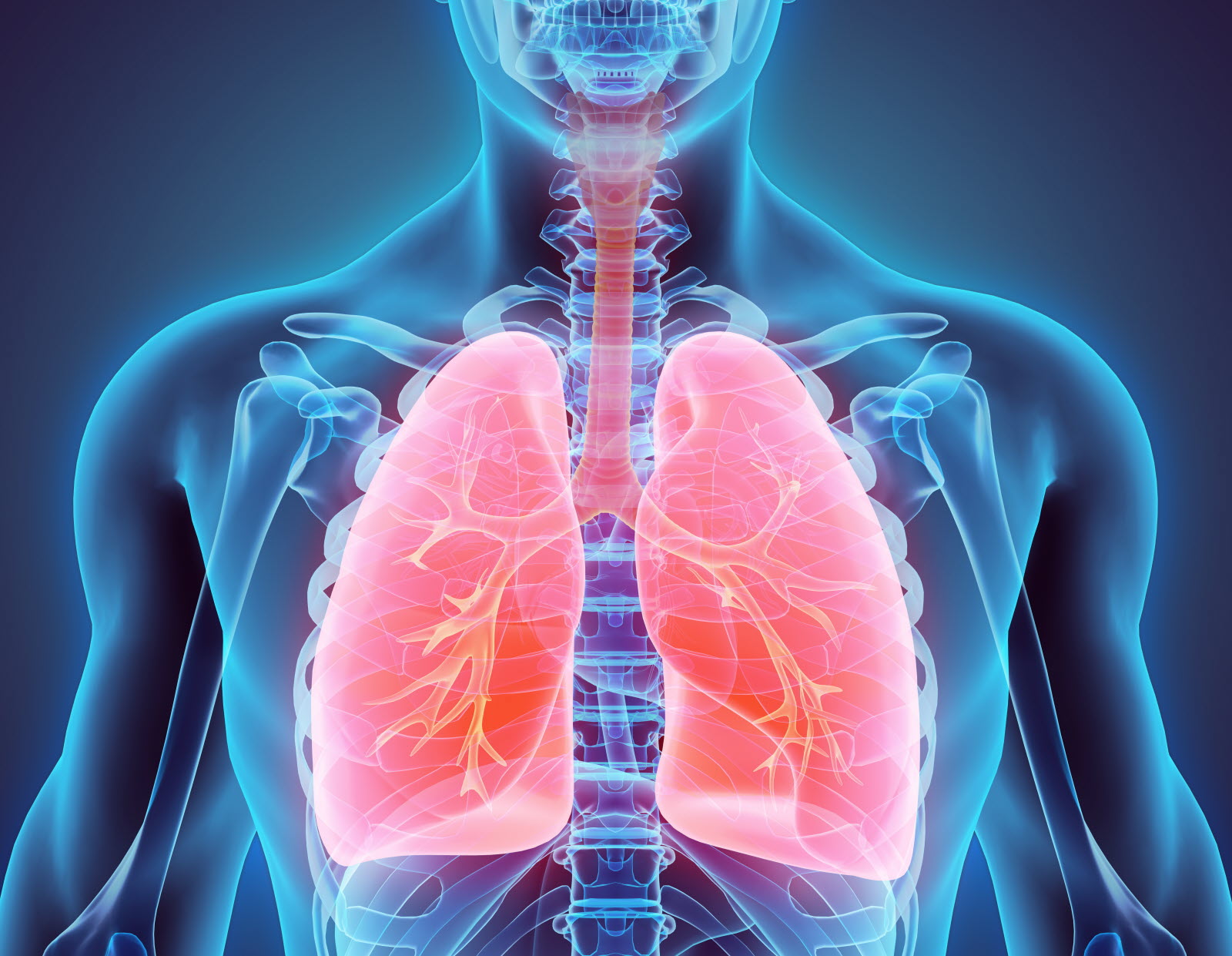
Les organes creux sont les principales cibles de la maladie car elle obstrue leurs canaux. Les bronches sont surtout touchées : lorsqu’elles sont bouchées par du mucus, elles présentent un risque d’infection car les bactéries ne sont pas évacuées convenablement. S’en suit alors une inflammation des tissus pulmonaires, leur destruction, une diminution progressive de la fonction respiratoire et à terme la mort du patient. Il se fait alors suivre par un kinésithérapeute dont la mission consiste à améliorer la clairance muco-ciliaire et à réduire l’infection pulmonaire. On utilise également des antibiotiques, des bronchodilatateurs, des anti-inflammatoires et des fluidifiants. La maladie se manifeste aussi par des troubles digestifs liés aux atteintes de l’intestin et du foie, et hépatiques liés aux atteintes rénales. C’est pourquoi les patients sont également suivis par des nutritionnistes. De plus, lorsque c’est possible, les médecins peuvent réaliser une greffe cœur-poumon ou poumon uniquement. Des atteintes au niveau génital sont également courantes, rendant 98% des hommes stériles. La femme reste fertile mais il lui sera difficile de tomber enceinte sans avoir recours à une insémination endo-utérine.
Article rédigé par Marie Ulmann
Bibliographie :
- https://www.pasteur.fr/fr/centre-medical/fiches-maladies/mucoviscidose
- https://www.inserm.fr/information-en-sante/dossiers-information/mucoviscidose
- https://www.vaincrelamuco.org/vivre-avec/la-mucoviscidose
Sources photographiques :
Comments are closed